USP Residual solvents
Organic solvents are often used in the manufacture or purification of drug substances, excipients or drug products but due to their potential toxicity, their absence/presence must be verified in the pharmaceutical products to ensure patient safety. For the finished product, the client may choose to test either all the individual components or the final finished product.
The United States Pharmacopeia (USP) method <467> provides detailed procedures for screening, confirmation and quantitation of residual solvents, including sample preparation and analytical conditions. Alternative methodologies may also be developed and used to demonstrate compliance with the defined solvent limits but will require validation of internal method for determining residual solvents rather than using the USP Residual Solvents <467> method.
Gas chromatography (GC) coupled with headspace (HS) sampling technique and flame ionization detection (FID) as detector is the analytical method specified in USP<467> for this application, as most of the target compounds are organic solvents with relatively low boiling points and good thermal stability.
The USP is harmonized with limits set out in the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Harmonised Tripartite Guideline for Residual Solvents Q3C approach for the classification of residual solvents. These solvents are evaluated for their possible risk to human health and are placed into one of three classes based on their toxicity data and their environmental impact as shown in Table 1.
USP Residual Solvents <467> is separated into three classes based on their potential toxicity level.
Table 1: Residual Solvents’ Classification and Their Risk Assessments
| Residual Solvents Classification | Risk Assessment |
| Class 1 (Solvents to be avoided) | Known human carcinogens |
| Strongly suspected human carcinogens | |
| Solvents particularly known to have environmental hazard properties | |
| Class 2 (Solvents to be limited) | Nongenotoxic animal carcinogens or possible causative agents of other irreversible toxicity, such as neurotoxicity or teratogenicity |
| Solvents suspected of other significant but reversible toxicities | |
| Class 3 (Solvents with low toxic potential) | Solvents with low toxic potential to humans; no health-based exposure limit is needed |
Solvent Limits
To ensure safety, final products are tested to assess whether the solvents used during the manufacturing processes have been efficiently removed or, if still present, their concentration is within the accepted limits.
Information Needed for Residual Solvent Testing
What the sample type is? Water or water-insoluble?
Residual solvents used in the manufacturing process (if known)?
For identification, Procedure A and B to be used to identify unknowns against reference standards as a Limit Test
For quantification purposes and known solvents likely to be present, Procedure C is used, although verification may be required to ensure the reliability of the compendial procedure.
USP<467> requires a minimum of 0.5g per release testing. For verification purposes, an additional 5-10g may be required on the complexity of the verification requirements.
Instruments Available
Perkin Elmer GCs with Turbomatrix HS-40 with FID
Thermo Scientific Trace1310 GC with ISQ MS and FID (Liquid Injection)
Thermo Scientific Trace1310 GC with TSQ8000 Evo MS/MS and FID with TriPlus RSH (Liquid Injection, Headspace, SPME)
ISQ MS and TSQ MS/MS can also be used to unequivocally identify unknown residual solvents, if required.

Clarus 580C GC with Liquid Autosampler (Left) and Turbomatrix HS40 (Right)

Trace 1310GC (Right) with TSQ8000 Evo (Left) and TriPlus RSH autosampler (Top)
USP Particulate matter in injections
Foreign particulate matter (FPM) in parenteral injections or infusions is defined as mobile, undissolved particles, other than gas bubbles, unintentionally present in solutions1. The presence of foreign matter can arise through the manufacturing or preparation process and can have serious consequences for the quality and effectiveness of the injections and patient safety.
There are a number of USP regulations for FPM which govern the limits on the particle size and particle count within a given product. One of the primary USPs for injections is USP <788> Particulate Matter in Injections. This specifies the acceptance criteria for the number of particles present in the majority of small- and large-volume parenteral injections. In particular the focus is on sub-visible particles, i.e., particles of ˂100µm in size. Two classifications of particle size are presented, dependant on the injection volume, according to Table 1 below.
Table 2. Acceptable particle content in parenteral injections according to USP<788>
| Injection volume | Test sample | Particle content ≥ 10µm | Particle content ≥ 25µm |
|---|---|---|---|
| Greater than 100 mL | Single container | Fewer than 25 particles/mL | Fewer than 3 particles/mL |
| Less than 100 mL | Single 25 mL container or 25 mL from at least 10 containers combined | Fewer than 6000 particles/container | Fewer than 600 particles/container |
The preferred method given in USP <788> for testing the presence of particulate matter in parenteral samples is a Light Obscuration Particle Count Test. A particle suspension flows past a laser beam, resulting in a change to the light intensity measured by the detector. From the profile of the scattered light, the particle size can be determined.
At Concept Life Sciences, the APSS 2000 is used for carrying out these tests. The APSS2000 is a light obscuration particle sizer with a parenteral autosampler. It is designed to meet all USP, EP and JP requirements, supporting a range of liquids and suspensions. It can be used for determining the particle count in a sample over the range of particle sizes from 2-125µm and is capable of measuring sample volumes from 0.2ml to 1 liter.
USP and Elemental impurities
Navigating the regulatory framework for elemental impurities can be challenging. There have been multiple changes to the guidance documents in recent years resulting in a step-change away from classical wet chemistry methods, with the elimination of the old USP <231>, towards specialized, element specific testing detailed in general chapters <232> and <233>.
Both of these general chapters, as well as the harmonized Ph. Eur. equivalents, relate to ICH Q3D and require the targeted assessment of up to 24 elements. The analytes to be included in your elemental impurities assessment depends is dependent on a number of risk areas, examples of which are highlighted below.
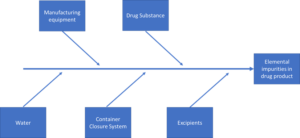
The intended administration routes (i.e., oral, parenteral or inhalation) and maximum daily dose are critical to identifying the correct limits to apply to ensure your method is fit for purpose. The maximum permitted daily exposure (PDE) is summarised for each element in Table 3.
Table 3: Maximum PDE µg/day
| Element | Class | Oral PDE | Parenteral PDE, | Inhalation PDE, |
|---|---|---|---|---|
| µg/day | µg/day | µg/day | ||
| Cadmium | 1 | 5 | 2 | 3 |
| Lead | 1 | 5 | 5 | 5 |
| Arsenic | 1 | 15 | 15 | 2 |
| Mercury | 1 | 30 | 3 | 1 |
| Cobalt | 2A | 50 | 5 | 3 |
| Vanadium | 2A | 100 | 10 | 1 |
| Nickel | 2A | 200 | 20 | 5 |
| Thallium | 2B | 8 | 8 | 8 |
| Gold | 2B | 100 | 100 | 1 |
| Palladium | 2B | 100 | 10 | 1 |
| Iridium | 2B | 100 | 10 | 1 |
| Osmium | 2B | 100 | 10 | 1 |
| Rhodium | 2B | 100 | 10 | 1 |
| Rubidium | 2B | 100 | 10 | 1 |
| Selenium | 2B | 150 | 80 | 130 |
| Silver | 2B | 150 | 10 | 7 |
| Platinum | 2B | 100 | 10 | 1 |
| Lithium | 3 | 550 | 250 | 25 |
| Antimony | 3 | 1200 | 90 | 20 |
| Barium | 3 | 1400 | 700 | 300 |
| Molybdenum | 3 | 3000 | 1500 | 10 |
| Copper | 3 | 3000 | 300 | 30 |
| Tin | 3 | 6000 | 600 | 60 |
| Chromium | 3 | 11000 | 1100 | 3 |
To add to the complexity, you may choose to validate a method at 30% of the permitted daily exposure in order to provide the data for your risk assessment in your submission dossiers which would serve the purpose of limiting testing requirements for your final product futuristically, ultimately saving time and resources over the lifecycle of your product without compromising on quality and safety. We have extensive experience within identifying the tests required, designing validation and providing the data and ensuring compliance with the regulations.
Whether part of a full GMP manufacturing campaign placed with us, a standalone analysis to provide robust analytical data for your risk assessment or a reliable provider for your release test, we have extensive analytical expertise, a comprehensive range of preparation equipment and high specification analytical instruments required to support.
- ICP-OES, Thermo Scientific iCAP-6500 Duo, with Qtegra Software
- ICP-MS, Agilent 7900, with Masshunter Software
USP Assessment of extractables associated with pharmaceutical packaging / delivery systems
Biocompatibility testing includes extractables and leachables (E&L) phases which investigate substances (organic and inorganic) that could potentially be released to a patient or user from medical devices, pharmaceutical production, and pharmaceutical packaging.
Extractables are defined as contaminants that can be extracted from plastics (and other materials) under exaggerated conditions such as using a volatile non-polar solvent at accelerated temperatures for 72 hours for example.
Leachables are those substances that are likely to be present in the product due to their close contact with a production system (and storage), container, and delivery system, and which might be leached out during normal usage.
The aim of an E&L study is to identify and quantify chemical species to allow an informed toxicological risk assessment and to support a biological evaluation, including Analytical Evaluation Threshold (AET).
USP <1663> and <1664> are key guidelines for such studies, detailing:
- Background, Concept and Theory
- Study Design
- Extraction Methodologies
- Characterization Approaches
- Chromatography (LC, GC, IC)
- Spectroscopy and Spectrometry
- Wet Chemistry (TOC, ROI, pH)
- Elemental
- Analytical Method Requirements
- Validation Requirements
- Extractables-Leachables Correlations
- Specification and Acceptance Criteria


